Death Domains: not exactly like Malfoy Manor

Death eater mask from the Harry Potter movies (Warner Bros). Totally unrelated to the article, but the term “death domain” somehow reminded me of the Death Eaters.
Subject areas: Cell Biology, Microbiology
Vocabulary:
apoptosis – programmed cell death. An orderly progression of events gradually breaking down internal cell components. This is regulated by a series of enzymes known as caspases. A signal causes the activation of one set of caspases. These make activate a bunch of other caspases. Each of those caspases activates yet another groups of caspases, and so on. Each step generates more active caspases than the one before. This is known as the caspase cascade. The caspases, in addition to activating other caspases, are enzymes that cut apart proteins, so their job is to take apart the cell from the inside to prepare for a clean and orderly cell death. As apoptosis progresses, the broken-down parts are carefully packaged so as not to leak out and affect neighboring cells.
immunoblot – also known as “Western Blot”. A sample of proteins is run on an electrophoresis gel, which separates them by size. These are then transferred (“blotted”) onto a paper-like membrane. This membrane is then incubated in a solution containing an antibody that will specifically bind to a particular molecule or part of a molecule. The antibody is attached to an enzyme that generates either a colored product or fluorescence. Either way, a picture can be taken (as seen in the second figure below).
The article:
Pearson, J.S. et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501:247-251 (2013) doi:10:1038/nature12524
First, I just need to say how fun it must be to write a title for a research article that says “blah blah blah ANTAGONIZES DEATH blah blah.” Personally I would have been tempted to add “and lived to tell about it” to the end of their title, but I suspect the journal’s editors would cut it out anyway.
In any case, this is paper with lots of unintuitive (for the lay person) photos of electrophoresis gels and complex graphs, but I wanted to write about it to walk you through the important points and ideas, to see the logic and thought process, and perhaps make sense of those figures.
What They Knew
There are two general ways that a cell can die: clean and messy. Messy is very messy: the dying cell gets leaky and starts spewing its insides out to the surrounding microenvironment, getting the stuff on neighboring cells, sometimes causing them to get sick too. On the other hand, a clean cell death, more scientifically known as apoptosis or programmed cell death, is very tidy and compartmentalized. It breaks things down internally, wraps them up in membrane-enclosed packages, and finally the cell folds itself into a bunch of such packages for the body’s macrophages to pick up and dispose of.
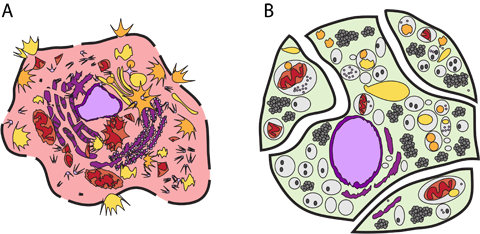
(A) Necrosis. The cell membrane has become leaky and is spewing its insides into its environment. (B) Apoptosis. Even as it breaks down its internal structures, it compartmentalizes and sub-compartmentalizes to keep everything cleanly packaged. Figure from Cells: Molecules and Mechanisms.
There are receptors on many cells called death receptors. When a specific signal is sent out and then sticks to the receptor, the death receptor activates. Activation, in this case, means changing its internal shape so that it can interact with specific proteins that will unleash the cascade of events that is apoptosis. Paradoxically, cell death can actually be a way to protect the organism from harm. By killing abnormal or infected cells before they can affect other parts of the body, a disease can be limited and even eliminated.
This paper focuses on certain pathogenic bacteria that invade the gut, and inject a number of bacterial proteins into the cells of the host animal’s gut lining. Surprisingly, these host cells do not die, rather they found that the host cells stayed alive despite the infection. It appears that this is good for the bacteria because they hang onto the gut cells and reproduce and shed into the feces for a long time, which is an avenue for spreading the infection to new hosts. If the host cells die upon infection, the bacteria do not have much time to reproduce before being shed from the host completely, and therefore limits infection to both the host and other organisms.
They knew that a protein from pathogenic bacteria, NleB1, seemed to inhibit a part of the apoptotic pathway from TNF (Tumor Necrosis Factor). Normally, if tumor necrosis factor binds to the TNF death receptor on a cell, it activates the apoptosis program to kill the cell. What they wanted to find out is exactly what NleB1 binds to, in order to figure out the specifics of how it interferes with cell death.
What They Did
The first step was to try to find cellular targets for NleB1. They used a “yeast 2-hybrid” system to do this, which sounds fancy (and was quite a technological breakthrough) but is a simple concept. What follows is a grossly oversimplified explanation. One group of yeast are transformed with DNA to make NleB1 (which they normally don’t). Another group of yeast are transformed with random chunks of DNA taken from a gut lining cell. Those yeast will make whatever bits of proteins are encoded on those pieces of DNA. So to reiterate, one population of yeast making NleB1, another population of yeast that are eaching making some random potential target for NleB1. They are mixed up and plated, and the researchers look for interaction between the NleB1 yeast and whatever other yeast carries a binding partner for it. The yeast are designed to show a signal (like a color change) if there is interaction.
After a few rounds of this, they found that NleB1 bound to FADD, a death-domain-containing protein that acts as an adaptor between a death receptor and the proteins that eventually initiate apoptosis. They also found that it bound to RIPK1, and enzyme, and to TRADD, another adaptor, both of which are known to bind to the TNF death receptor, and are probably involved.
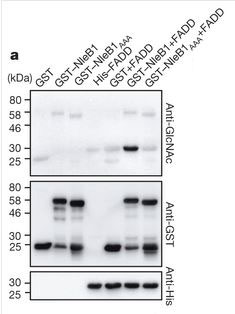
This is an immunoblot (see vocabulary) showing sugar-tagging enzyme activity by NleB1. The top panel shows staining for the sugar attachment. You can see that it is only in the column containing FADD and NleB1. All other columns, which include NleB1 by itself, FADD by itself, control protein, and mutated non-functional NleB1 with FADD, show no active enzyme activity.
So now they know the targets, but they still don’t know exactly what NleB1 does to prevent apoptosis. From work on a close relative of NleB1, they suspected that NleB1 could be an enzyme that added a molecular tag of sugars to a protein. Could this be how it disrupts apoptosis? They tested and confirmed that NleB1 could in fact stick a tag onto FADD. They then tried to figure out if this tag was in a place that could actually affect function. It turns out that the tag location is exactly in the middle of an area that is crucial for assembling the protein complex that helps TNF receptor signal apoptosis.
What is next? This just showed that NleB1 put down a tag in a place that could disrupt apoptosis, but doesn’t show it must be the case. So, they created a mutant NleB1 that had just one little mutation that turned off the tagging ability. The non-tagging NleB1 did not inhibit apoptosis (or at least the first caspase in the cascade).
Now they decided to test their ideas in vivo. They knew that FADD and TRADD are part of a complex with FAS, and they could obtain mice that were engineered with a defective FAS. They then infected normal mice and FAS-defective mice with Citrobacter rodentium, which is a mouse pathogen that uses NleB1. Basically, having the defective FAS should be like giving the bacteria a head start, and in fact, the FAS-defective mice were in much worse shape after infection, with serious watery diarrhea and much longer time of infection (the normal mice eventually cleared the bacteria out).
What They Know Now
The researchers were able to show quite convincingly that NleB1 is an important part of the pathogenicity of a type of gut bacteria that includes human pathogens like Salmonella, Shigella, and some strains of E. coli. They also showed that its cellular targets are adaptor and effector proteins that have “death domains” that interact with the tumor necrosis factor receptor. They showed that it deactivates these targets by attaching a multi-sugar tag onto them. And finally, they showed that not only was all of this the case in cell culture and biochemical experiments, but their results were predictive for live animal models.
Hopefully that gives you a sense of the mindset and methodical logic that characterizes many of us who are or have been professional research scientists.
No comments
Be the first one to leave a comment.